So, a new ctenophore genome has just been published in Nature (Moroz et al., 2014), it makes some extraordinary claims, and my resident palaeontologist/web-buddy Dave Bapst wants my opinion 😉
Given that I already planned to have an opinion about the first ctenophore genome back in December (Ryan et al., 2013) and miserably failed to finish the post… the temptation is just too strong. (That thesis chapter draft in the other window of MS Word wasn’t going to be finished today anyway >_>)
Whatever I might seem from words on the internet, I’m not some kind of expert on phylogenetics, so I’m going to use a crutch. I had this idea back when I first read Ryan et al. (2013), because I remember thinking that it was written almost as if Nosenko et al. (2013) had never happened, and I’d really liked Nosenko et al. (as you can guess from the word count of this post), so I was mildly indignant about that. The Nosenko paper is going to be my crutch. (No offence to Hervé Philippe and friends, but there are only so many papers I’m going to reread for an out of the blue blog post 😉 )
Although I’m obviously not writing a public post specifically for a phylogeny nut, I may get somewhat technical, and I’m definitely going to get verbose.
***
Ctenophores. Comb jellies, sea gooseberries, Venus girdles. They are floaty, ethereal, mesmerizingly beautiful creatures, and I have it on good authority that they are also complete pains in the arse.
Here’s some pretty pictures before it gets too painful 😉 Left: Mnemiopsis leidyi from Ryan et al. (2013); right: Pleurobrachia bachei from Moroz et al. (2014). And a bonus video of a Venus girdle making like an ancient nature spirit. I could watch these beasties all day.

Venus from Sandrine Ruitton on Vimeo.
The problem(s)
And now, the pain. Let’s pull out my trusty old animal phylogeny, because the question marks are once again highly appropriate. (Also, I’m hell-bent on breaking your bandwidth with PICTURES.)
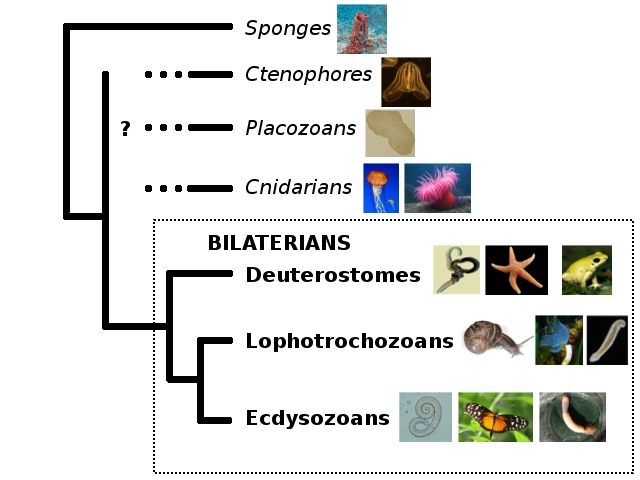
Ryan et al. (2013) helpfully have a figure distilling the ideas people have had about those question marks so far:

Bi = bilaterians, Cn = cnidarians, Ct = ctenophores, Tr = Trichoplax, and Po = sponges (Porifera).
I say “helpfully,” but it’s not all that helpful after all, since pretty much every possible configuration has been proposed. Why is this such a difficult question? Here’s a quick rundown of the problems Nosenko et al.’s study found to affect the question marks:
- Fast-evolving protein sequences – these can cause artefacts because too much change overwrites informative changes and creates chance similarities. Excluding faster-evolving sequences from the analysis changes the tree.
- Sequence data that don’t conform to the simplifying assumptions of popular evolutionary models – again, this can result in chance similarities and artefacts, and using a poorer model replicates the effects of using less ideal sequences.
- Long-branched outgroups – these are the non-animal groups used to place the root of animals. The more distant from animals and less well-sampled the outgroup, the longer the branches it forms, which can attract fast-evolving animal lineages towards the root. In Nosenko et al.’s analyses, even the closest outgroup seemed to cause problems, and removing the outgroup altogether made the conflicts between different models and datasets disappear completely – but this isn’t exactly helpful when you’re looking for the root of the animal tree!
The problem with ctenophores in particular is illustrated by this one of Nosenko et al.’s trees, made from one of their less error-prone datasets:

The ctenophore branch is not only longer overall than pretty much any other in the tree; its length is also very unevenly distributed between the loooong history common to all species and the short unique lineage of each individual species. That is bad news. And it may stay that way forever, because the last common ancestor of living ctenophores may genuinely be very recent, so there’s no way to divide up that long-ass internal branch without a time machine.
Round 1: Nosenko vs. Ryan
In fairness, the Mnemiopsis genome team probably didn’t have a whole lot of time to specifically deal with Nosenko et al.’s points (OTOH, none of those individual points were truly new). The Nosenko paper came out in January 2013, and the Mnemiopsis genome paper was received by Science in July of the same year – I imagine most of the data had been generated way before then, and you can’t just redo all your data analysis and rewrite a paper on short notice.
I’m still going to view Ryan et al. (2013) in the light of Nosenko, because regardless of the genome team’s ability to answer them, some of Nosenko et al.’s points are very relevant to the claims they make. Their biggest claim, of course, being that ctenophores are the sister group to all other animals.
In Nosenko et al.’s experiments, this placement showed up in trees where faster-evolving genes, poorer models or more distant outgroups were used, but not when the slowest-evolving gene set was analysed with the best models and the closest outgroup.
Ryan et al. acknowledge that “supermatrix analyses of the publicly available data are sensitive to gene selection, taxon sampling, model selection, and other factors [cite Nosenko].” Their data are obviously sensitive to such factors. In fact, they behave rather similarly to what I saw in the Nosenko study.
Ryan et al. used two method/model combinations – one of the models was the preferred CAT model of Nosenko et al., and the other was the OK but not great GTR model that CAT beat by miles in terms of actually fitting Nosenko et al.’s data. (Caveat: in the genome paper, the CAT and GTR models were used with different treebuilding methods, so we can’t blame the models for different results with any certainty.) Also, they analysed the data with three different outgroups.
And guess what – the ctenophores-outside-everything tree was best supported with (1) the GTR model, (2) the more distant outgroups. There is not much testing of the effect of gene choice – there were two different data sets, but they were both these massive amalgamations of everything useable, and they also included totally different samples of species.
However, here comes another nod to Nosenko et al. and all the other people who advocated trying things other than “conventional” sequence comparisons through the years. Provided you can securely identify genes across different organisms, you can also try to deduce evolutionary history based on their presences and absences rather than their precise sequences. This is not a foolproof approach because genes can be (commonly) lost or (occasionally) picked up from other organisms, but it is often regarded as less artefact-prone than sequence-based trees.
But does it help with ctenophores? Like the GTR model-based sequence trees, the tree based on gene presence/absence (you obviously need complete genomes for this!) supports ctenophores being the outsider among animals:

My problem with this? Note what else it supports. The white circles indicate groupings that this method had absolutely no doubt about. And these groupings include things that frankly sound like abject nonsense. Here’s one annelid worm (the leech Helobdella) sitting next to a flatworm, while another annelid worm (Capitella) teams up with a limpet right next to a chordate. If anything, that is more controversial than the placement of ctenophores, because we thought we had it settled!
So if we’re concluding that ctenophores are basal to all other animals, why aren’t we also making a fuss about the explosion of phylum Annelida? Surely, if this method gives us strong enough conclusions to arbitrate between different sequence-based hypotheses about ctenophores, it’s strong enough to make those claims too. The cake can’t quite decide if it’s being eaten, I think.
I’m not sure what to think about the sequence trees. I’m far more confident about the presence/absence one. Maybe I’m just demonstrating the Dunning-Kruger effect here, but I’m not buying that tree for a second.
Overall verdict?
Not convinced. Not by a long shot.
Round 2: Nosenko vs. Moroz
The Pleurobrachia genome took me completely by surprise. I’d known Mnemiopsis was sequenced since Ryan et al. (2010). (Three years. Can you imagine the twitching?) I had no idea this other project was happening, so I nearly fell off my chair when Nature dropped it into my RSS reader yesterday. Another ctenophore genome – and another one that supports ctenophore separatism? (This hypothesis is becoming strangely popular…)
Bonus: it’s not just a genome paper, it also describes the transcriptomes of ten different ctenophores. Transcriptomes, the set of all active genes, are a little bit easier to sequence and assemble than genomes, and if you’re thorough they’ll catch most of the genes the organism has, so they can be almost as good for the analysis of gene content.
Which they kind of don’t do properly. There is a discussion of specific gene families that ctenophores lack – including many immune- and nervous system-related genes – but that’s not exactly saying much given that we know even “important” genes can be lost (case in point: the disappearing (Para)Hox genes of Trichoplax). The fact that ctenophores seem to completely lack microRNAs is interesting, but again, it doesn’t mean they never had them. Sponges do have microRNAs but don’t seem to be nearly as big on them as other animals.
As for the global analysis of gene content – I had to chase down a reference (Ptitsyn and Moroz, 2012) to understand what they actually did. As far as I can tell, there is no phylogenetic analysis involved – they just took a tree they already had, and used this method to map gene gains and losses onto that tree. Which is cool if you’re fairly sure about your tree, but pretty much meaningless when the tree is precisely the question. The Mammal is disappointed.
One of the problems with listing genes that aren’t there or don’t work in the “expected” way in ctenophores is that even if they’re not outside everything else, it’s still a distinct possibility that these guys branched off from our lineage before cnidarians did. For example, the Pleurobrachia paper spends a lot of time on “nervous system-specific” genes like elav missing or not being expressed in neurons, and common neurotransmitters like serotonin not being used by ctenophores.
But, assuming that the tree of animals looks something like (sponges + (ctenophores + (cnidarians + bilaterians))), we wouldn’t expect ctenophore nervous systems to share every property that cnidarians and bilaterians share. Remember: (1) sponges don’t have nervous systems, so they’re not much use as a comparison, (2) cnidarians + bilaterians had a longer common ancestry than either did with ctenophores. Genes possessed by sponges PLUS cnidarians and/or bilaterians but missing from ctenophores are more suggestive, but only if you can demonstrate that they weren’t lost. (We’re kind of going in circles here…)
The other problem is that pesky last common ctenophore ancestor. If it really is very recent, then taking even all living ctenophores to represent ctenophore diversity is like taking my close family to represent human diversity. Just like my family contains pale-skinned, lactose tolerant people, it is entirely possible that this lone surviving ctenophore lineage possesses (or lacks) important traits that aren’t at all typical of ctenophores as a whole. Ryan et al.’s supplementary data are clear that at least the Mnemiopsis genome is horribly scrambled, all trace of conserved gene neighbourhoods erased from it. That’s not exactly promising if you’re hoping for “trustworthy” animals.
The actual phylogenetic trees in Moroz et al. (2014) seem to follow an approach of throwing AAAALLL the genes at the problem. The biggest dataset contains 586 genes, compared to 122 in Nosenko et al.’s largest collection, and there is not much filtering by gene properties other than “we can tell what it is”. I have no idea how the CAT + WAG model they used compares to CAT or WAG or GTR on their own; unfortunately, the Nosenko paper doesn’t test that particular setup and this one doesn’t do any model testing. Moroz et al.’s supplementary methods claim it’s pretty good, cite something, and I’m not gonna chase down that reference. (Sorry, I’ve been poring over this for four hours at this point).
Interestingly, the support for ctenophores being apart from other animals increases when they start excluding distant outgroups. The only time it’s low is when they add all ten ctenophores and use fewer genes. Hmm. This is where I would like to hear some real experts’ opinions, because on the face of it, I can’t pinpoint anything obviously wrong. (Other than saying that chucking more genes at a problem tree is perfectly capable of making the problem worse)
TL;DR version: While I’m generally underwhelmed by the gene content stuff, I literally have no idea what to think about the trees.
I’m banking on the hope that someone will do.
***
And… I think that is all the opinion I’m going to have about ctenophores for a long time. Lunch was a long time ago, my brain is completely fried, and I’m not sure how much of the above actually makes sense. To be clear, I don’t really have a horse in this race, though I’d really like to know the truth. (Fat chance of that, by the looks of it…) I think I’m going to need a bit more convincing before I stop looking sideways at this idea that ctenophores are further from us than sponges. If anything is clear from recent phylogenomics papers, it’s that what data you analyse and how you analyse them makes a huge difference to the result you get, and this is happening with data and methods where it’s not necessarily easy to dismiss an approach as clearly inferior.
It’s a mess, damn it, and I’m not qualified to untangle it. Urgh.
***
References
Moroz LL et al. (2014) The ctenophore genome and the evolutionary origin of neural systems. Nature advance online publication, 21/05/2014; doi: 10.1038/nature13400
Nosenko T et al. (2013) Deep metazoan phylogeny: When different genes tell different stories. Molecular Phylogenetics and Evolution 67:223-233
Ptitsyn A & Moroz LL (2012) Computational workflow for analysis of gain and loss of genes in distantly related genomes. BMC Bioinformatics 13:S5
Ryan JF et al. (2010) The homeodomain complement of the ctenophore Mnemiopsis leidyi suggests that Ctenophora and Porifera diverged prior to the ParaHoxozoa. EvoDevo 1:9
Ryan JF et al. (2013) The genome of the ctenophore Mnemiopsis leidyi and its implications for cell type evolution. Science 342:1242592