When I discussed sponge microRNAs last week, I said deep animal phylogeny was difficult. Quite fortuitously, another paper went online recently that explores exactly this difficulty (Nosenko et al., 2013). Following on from the microRNA post, I’ll use this paper as an excuse/guide to discuss the tangled relationships of animals.
First of all, let’s recap the problem. My trusty old family tree of animals just so happens to be an excellent illustration:
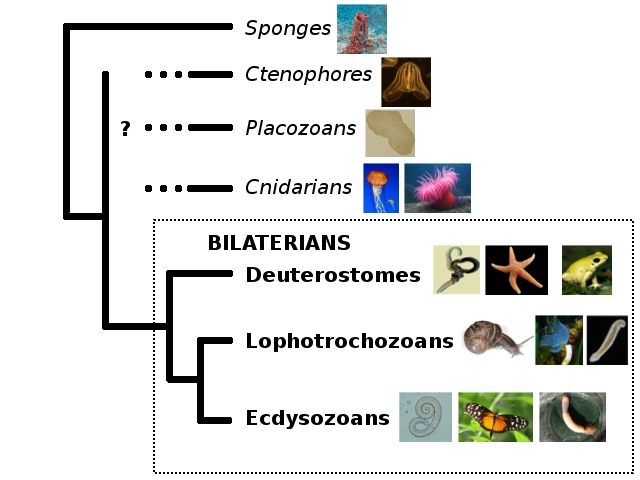
When I first made this tree to explain what the hell I was talking about re: the Cambrian creature Nectocaris, I put in some question marks mostly out of laziness. To illustrate why the “old” Nectocaris didn’t make sense, I only needed the relationships of bilaterians among themselves. Everything outside the Bilateria was irrelevant to the little creature’s mystery, so I decided to forgo reading up on them and stay on an uninformed fence.
But, in fact, said fence is not just my half-arsed perch. I appear to share it with an entire, very much whole-arsed field. While now there’s a reasonable agreement over ecdysozoans and deuterostomes and all that jazz, the non-bilaterians still wander all over the place depending on how you do your analysis. Nosenko et al. cite a number of recent large-scale studies, and point out that they totally fail to agree where to put poor Trichoplax and jellies of various kinds. The other thing they fail at is deciding how many branches sponges actually represent (the problem the microRNA study I discussed tried to tackle). To illustrate the extent of the chaos, I sketched the phylogenies six recent studies cited by Nosenko and colleagues came up with (sponge lineages are marked by dots):

Remarkably, all six studies agree on the basic deuterostome-ecdysozoan-lophotrochozoan arrangement inside Bilateria in spite of using different sets of bilaterian species. In contrast, the non-bilaterian animals – sponges of all kinds, cnidarians, ctenophores and Trichoplax – appear in pretty much every conceivable configuration.
A plethora of pitfalls
Why? What makes these questions so difficult that datasets made of 100+ genes from dozens of species representing all major animal groups and using the best available methods have this much trouble answering them?
Time is probably not the issue, or at least not in the simple sense of “it all happened too long ago”. The Nosenko paper brings up the example of fungi, which are roughly as ancient (or, in the context of all living things, as young) as animals. Studies that tried to use the exact same set of genes to analyse the relationships within each group could apparently produce a nice clear tree for fungi. Animals? A whole lot of noise.
Perhaps the “tree” of animals is really more like Rokas and Carroll’s (2006) evolutionary bushes, with its base branching so quickly that genes didn’t have time to accumulate many informative changes between one split and the next. Perhaps it even happened so fast that ancient within-species sequence variation was carried through several such events, resulting in what population geneticists call incomplete lineage sorting, a situation where the history of genes is not the same as the history of species.
Perhaps we haven’t got a good enough sample of genes, animals, or both.
If early animal evolution was bush-like, only a large amount of good data has any hope of accurately resolving how it went. But finding suitable genes for phylogenetic analysis is not easy. They have to be known in all of our species. They should have unambiguous identities so we know we’re actually comparing the same gene across species. They should evolve slowly enough that chance hasn’t had time to wash away their records of relatedness.
Likewise, picking suitable species can be difficult. Aside from the availability of sequences, the two greatest problems are taxon sampling and long branches. Good taxon sampling means covering the diversity of a group. So for example, if you have to pick three vertebrates, you don’t want them all to be mammals. A mammal, a shark and, say, a bony fish would be a much more representative sample.
Long branches are the bogeyman of phylogenetics. “Long” here means many evolutionary changes compared to other lineages in your sample. Similarities in gene/protein sequences are not always due to shared ancestry: because there’s a limited number of letters in the DNA and protein alphabets, sometimes they happen just by chance. If you have two unusually long branches, they might have a lot of these chance similarities, many more than either of them shares with its true relatives by common ancestry. Some of the newer changes might also have overwritten the older similarities linking them with their real families, a problem known as saturation. The overall outcome is that long branches attract each other.
Last but not least, perhaps the assumptions we put into our analyses don’t actually fit the data. All phylogenetic analyses are based on a model of evolution. For molecular data, these models specify, for example, how likely different sequence changes are, and which bases or amino acids are commonest and rarest. All analyses also need a way of picking the best tree, which range from simply choosing the one with the fewest changes to choices based on complicated probability theory. Sometimes, models and methods still work reasonably well when their assumptions are violated, but, as you might expect, counting on that is generally a stupid idea.
Nosenko et al. (2013) come to the conclusion that the issue of non-bilaterian animal phylogeny is plagued by pretty much the whole package.
Dissecting the Problem
First, studies may have increased the size of their datasets by incorporating less than ideal genes. To test the effect of gene sampling, Nosenko et al. (2013) divided their collection of 122 genes into two parts. One consisted of genes involved in protein synthesis, mostly genes encoding ribosomal proteins, which all evolve very slowly. The other was a mixed bag of non-ribosomal genes with all sorts of functions and evolutionary rates.
Perhaps not surprisingly, the latter set displayed a much higher level of saturation. Accordingly, when they analysed the ribosomal dataset with models of evolution that are more prone to errors due to saturation, they got the same trees they’d seen using more accurate models on the non-ribosomal data. Clearly, saturation, gene and model choice are affecting the answers they’re getting, and they are all problems that would affect your average phylogenomic study.
Second, the authors found every indication of a serious long-branch problem. In most phylogenetic trees, the longest branch is the outgroup. Outgroups are organisms outside your group of interest (the ingroup). Similarities between the outgroup and members of the ingroup are likely to have evolved before the origin of the ingroup, therefore they can be used to locate the root of the ingroup tree. However, outgroups are rarely sampled as well as ingroups, hence they tend to form long branches, making them a liability.
In the case of animals, removing the outgroup cleared the disagreements between the different gene sets, demonstrating that some of them had been due to long-branch artefacts. (Of course, without an outgroup you don’t know which animal lineages split first, which makes this solution not much use at all for important evolutionary questions like what the common ancestor of all animals looked like.)
Likewise, using a more distant outgroup changed the trees considerably. Ctenophores are worth special mention here. When Dunn et al. (2008) placed these jellyfish-like creatures as the sister group to all other animals, it was an odd, unexpected result. Well, ctenophore genomes evolve ridiculously fast, and there’s a good chance that their position “way out there” is an artefact of that. In Nosenko et al.‘s analyses, they ended up in the Dunn position when the more saturated non-ribosomal data were used – or when the ribosomal dataset was analysed with a more distant outgroup. When everything possible was done to reduce long-branch issues, they stayed deep in the crown of the tree next to cnidarians.
Fourth, the assumptions of even the best evolutionary model don’t take into account an annoying property of protein sequences: their overall amino acid compositions can differ across lineages. Changing the entire makeup of an organism’s protein complement involves changes in evolutionary patterns that none of the models account for. Once again, those damned ctenophores are one of the problem taxa with “deviant” sequence compositions. (The even worse news is that the closest available outgroups also differ from typical animals in this respect.)
Fifth, taxon sampling is influencing what you get. For example, the more sponges Nosenko et al. included, the more support they got for sponges being a single lineage. Ctenophores probably also suffer from this problem. For one thing, they’re very poorly known in almost every way that is relevant to picking species for phylogenetic analysis.
For another, they may actually have an additional problem that is literally impossible to crack – phylogenetic analysis of ctenophores themselves and a look at their fossil record hint that most ctenophore lineages have died out, with existing species all coming from a relatively recent common ancestor. That would make the entire phylum incurably long-branched no matter how many living species you throw at your datasets!
And finally, the ribosomal dataset that was the least prone to long-branch artefacts and the most informative about the deepest branches in animal phylogeny comes with a big caveat: it’s not a random selection of genes. In fact, all of these genes are interacting parts of a single system, which means they might not evolve independently (in the statistical sense). Are they all affected by a common set of biases, and does it render them unsuitable for recovering the true history of animals? We don’t yet know.
Hope dies last…
Being the phylogeny nut that I am, I really enjoyed this dissection of a thorny problem. At the same time, the results are kind of depressing. (Especially if, like me, you’re interested in early animal evolution.) No matter how carefully you set up your analysis, biases lurk around the corner waiting to jump on you and destroy your conclusions. You have a choice between not knowing where to root the tree of animals and being screwed by the outgroup. Well-worn measures of statistical confidence can support contradictory hypotheses. Ctenophores are fucking hopeless.
Is there anything we can do about this conundrum? Nosenko et al. conclude their paper on a somewhat hopeful note. There are other methods in molecular phylogenetics than simple sequence comparison. Although they’ve been no more helpful so far than traditional sequence analysis, we’re getting more and more full genome sequences from all over the animal kingdom. There’s more to look at than ever. Perhaps, one day, we’ll find a tool that can trim this thorny beast of a bush (or bush of beasts?) into shape.
Meanwhile, the quandary of deep animal phylogeny stands as a reminder that science is not all-powerful. The universe is a puzzle, but we have no reason to assume that nature left us enough information to solve it all. Which, as far as I’m concerned, shouldn’t stop us from trying. 😉
***
References:
Dunn CW et al. (2008) Broad phylogenomic sampling improves resolution of the animal tree of life. Nature 452:745-749
Erwin DH et al. (2011) The Cambrian conundrum: early divergence and later ecological success in the early history of animals. Science 334:1091-1097
Nosenko T et al. (2013) Deep metazoan phylogeny: when different genes tell different stories. Molecular Phylogenetics and Evolution (in press), doi: 10.1016/j.ympev.2013.01.010
Philippe H et al. (2009) Phylogenomics revivew traditional views on deep animal relationships. Current Biology 19:706-712
Pick KS et al. (2010) Improved phylogenomic taxon sampling noticeably affects nonbilaterian relationships. Molecular Biology and Evolution 27:1983-1987
Rokas A & Carroll SB (2006) Bushes in the tree of life. PLoS Biology 4:e352
Schierwater B et al. (2009) Concatenated analysis sheds light on early metazoan evolution and fuels a modern “urmetazoon” hypothesis. PloS Biology 7:e20
Sperling EA et al. (2009) Phylogenetic-signal dissection of nuclear housekeeping genes supports the paraphyly of sponges and the monophyly of Eumetazoa. Molecular Biology and Evolution 26:2261-2274

4 thoughts on “Thornbushes”